Introduction
Misconducts in the STAP cell research
A new type of induced pluripotent cells, STAP cells, was reported in two Nature papers by Dr. Obokata in RIKEN CDB and the others in 2014. Immediately after the publication, misuses of the data in the papers were pointed out by several readers. RIKEN set up Fraud Investigation Committees for the STAP papers. After the interviews and investigations on the authors of the STAP papers, the comittees concluded that there was misconduct by Dr. Obokata including total 3 fabrications and 1 falsification of data [ref1]. For the other figures in the papers, Dr. Obokata did not show any original data even after repeated requests from the committee although most of the figures were created by her. The committee reported that "There are few records of experiments. There are some experiments that we cannot tell they really were conducted (althogh we cannot tell they were not conducted either). "
Concurrently with this investigation on the research process, the committe reviewed the results of the scientific investigation of the samples obtained in the STAP study. The scientific investigation team in RIKEN analized the genotypes of STAP stem cells [ref4]. They found that some of them were different from those of mice used in the study. An obtained genotype suggested that some STAP stem cells were derived from ES cells that had been established for other reseach purposes. They finally did whole genome seuquencings of the selected STAP related cells and mice that were related to the STAP study. Total 15 genomes were sequenced and the NGS data were pulished on public database [ref2].
The committee concluded from the analysis that STAP cells used in a teratoma expeiment, STAP cells used for ChIP-seq, STAP stem cells, and FI stem cells were all derived from pre-existing ES cells established in Wakayama lab in RIKEN CDB, where Dr. Obokata and Dr. Wakayama mainly did most of their STAP cell experiments. The detail of the investigation was published as Nature B.C.A. by the original scientific investigation team in 2015 [ref3]. Their conclusion that STAP cells are derived from ES cells is based on similarities found in the genome structure, which cannot be explained if they are from different sources.
My goal (ongoing)
My specific goal of this analysis of published NGS data is to confirm their results and their conclusion that STAP cells are derived from ES cells. My interest is whether the difference between the ES cells (e.g., FES1) and STAP cells are explained by mutation accumulation that occured during cell culturing. Although this explanation has been suggested in the Nature B.C.A. paper, it lacks the quantitative evaluation and modeling of this process.
Genome sequence (DRA002862 in DDBJ)
Instruments: Illumina HiSeq 2500
Library construction: TruSeq DNA PCR-free; 
Note: the sizes of the NGS reads are very big; .fastq - 67G byte, .fastq.gz - 20G byte, .fastq.bz2 - 15G byte
Note: There are 2 runs of NGS data for each cell. I don't know whether they are biological duplicates or technical duplicates.
Note: I can not share .bam files here because their sizes are too large (30-40GB) to store in servers I have rented.
FLS3 (ftp)
established at 1/31/2012-2/2/2012
STAP stem cell
| Run name | forward read | reverse read | VCF |
|---|---|---|---|
| DRR028632 | 1 (FastQC) | 2 (FastQC) | 320MB |
| DRR028633 | 1 | 2 |
CTS1 (ftp)
established at 5/25/2012 or 7/9/2012
FI stem cell
| Run name | forward read | reverse read | VCF |
|---|---|---|---|
| DRR028642 | 1 (FastQC) | 2 (FastQC) | 320MB |
| DRR028643 | 1 | 2 |
129/GFP ES (ftp)
unknown; found in Obokata's box in a freezer in Obokata lab.
Unknown type, possiblly ES cell
| Run name | forward read | reverse read | VCF |
|---|---|---|---|
| DRR028638 | 1 (FastQC) | 2 (FastQC) | 320MB |
| DRR028639 | 1 | 2 |
FES1 (ftp)
129B6GFP1 FES(male); frozen at 12/7/2005
ES cell
Transgene: Acr3-EGFP/CAG-EGFP derived from transgenic mouse created in Okabe lab. (strain B6).
estabilished from F1 (129X1 x B6N) mouse
| Run name | forward read | reverse read | BAM | VCF | IGV |
|---|---|---|---|---|---|
| DRR028646 | 1 (FastQC) | 2 (FastQC) | 320MB | View | |
| DRR028647 | 1 (FastQC) | 2 (FastQC) | stats. | 320MB |
FES2 (ftp)
129B6GFP2 FES(male); frozen at 12/7/2005
| Run name | forward read | reverse read | VCF | IGV |
|---|---|---|---|---|
| DRR028652 | 1 (FastQC) | 2 (FastQC) | 300MB | View |
| DRR028653 | 1 | 2 |
ntESG1 (ftp)
129B6F1G1; frozen at 8/3/2007; named ntES1 in DDBJ
nuclear transfer ES cell
Transgene: Acr3-EGFP/CAG-EGFP derived from transgenic mouse created in Okabe lab. (strain B6).
estabilished from F1 (B6N x 129+Ter) mouse
| Run name | forward read | reverse read | BAM | VCF |
|---|---|---|---|---|
| DRR028644 | 1 (FastQC) | 2 (FastQC) | stats. | 310MB |
| DRR028645 | 1 | 2 |
ntESG2 (ftp)
129B6F1G2; frozen at 1/20/2005; named ntES2 in DDBJ
| Run name | forward read | reverse read | BAM | VCF |
|---|---|---|---|---|
| DRR028650 | 1 (FastQC) | 2 (FastQC) | stats. | 300MB |
| DRR028651 | 1 | 2 |
Mouse reference genome (GRCm38)
strain C57BL/6J
file size: .fa = 4.4G byte, .fa.gz = 0.8G byte
$ wget ftp://ftp.ensembl.org/pub/release-74/fasta/mus_musculus/dna/Mus_musculus.GRCm38.74.dna.toplevel.fa.gz
Transgenes
see my analysis results
CAG-GFP
CAG-EGFP, CAGGS-EGFP, CX-EGFP
EcoRI-EcoRI fragment of PCR product (primers: 5'-ttgaattcgccaccatggtgagc-3' and 5'-ttgaattcttacttgtacagctcgtcc-3') using pEGFP-C1 as a template was introduced into pCAGGS vector (CAG + pA signal) (ref)
3.2-kb SalI/BamHI fragment from pCAGGS-EGFP was used when creating Tg mouse
EGFP sequence from pEGFP-C1 (Clontech)
CAG promoter (1.7kb) + EGFP (0.7kb) + rabbit beta-globin pA and vector (0.7kb) = 3.1kb
Arc-GFP
Acr3-EGFP (ref1)
mouse acrosin promoter and N-terminal coding region (2.4kb) + EGFP (0.7kb) + rabbit globin pA = 3.7kb (ref2)
XbaI-HindIII fragment was used when creating Tg mouse
The original paper (ref1) described that Acr3-EGFP transgene construct used bovine growth hormone polyA signal (bGH pA). But ref2 uses rabbit globin polyA signal perhaps from pCAGGS vector. My personal investigation indicated that FES1 genome has no bGH pA sequence but has rabbit polyA signal sequence.
Oct4-GFP
GOF18-EGFP
GOF mouse ;18-kb genomic fragment of mouse Oct4 gene containing Oct4 pomoter region **
Mouse SNPs Data
variations relative to the C57BL/6J mouse reference genome
Mouse Genomes Project (.bam files)(.vcf files)
from Methods from the analysis paper
For comparison to 129/Sv SNPs, we used commonly called SNPs in 129P2/OlaHsd (EMBL ENA: ERP000034), 129S1/SvImJ (EMBL ENA: ERS076385), and 129S5SvEvBrd (EMBL ENA: ERP000036) but not called in C57BL/6NJ (EMBL ENA: ERS076384) provided by the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/resources/mouse/genomes/).129P2/OlaHsd (.vcf.gz, 240M byte; .vcf, 1.9G; contains 6x10^6 SNPs)
$ wget ftp://ftp-mouse.sanger.ac.uk/current_snps/strain_specific_vcfs/129P2_OlaHsd.mgp.v5.snps.dbSNP142.vcf.gz
$ gzip -d ...vcf.gz $ bgzip ...vcf $ bcftools index ...vcf.gz
Update: I used a compiled version of VCF file obtained from ftp://ftp-mouse.sanger.ac.uk/REL-1303-SNPs_Indels-GRCm38/mgp.v3.snps.rsIDdbSNPv137.vcf.gz in later analyses. This file contains the alleles of all the inbed strains sequenced in Mouse Genomes Project.
Tools
see related file types
FastQC
A quality control application for high throughput sequence data
Babraham Bioinformatics; v0.11.5
written in Java
$ wget ... $ unzip ... $ sudo apt-get install default-jre
BWA
alignment via Burrows-Wheeler transformation (ref)
Heng Li; version 0.7.12-r1039
outputs .sam file
$ sudo apt-get install bwa
SAMtools/BCFtools
Tools for alignments in the SAM format
version 0.1.19-
for variation detections (SNPs, short INDELs, ...)
$ sudo apt-get install samtools
Later on, I used the latest version 1.3.1 for my analysis.
$ sudo apt-get update $ sudo apt-get install gcc $ sudo apt-get install libncurses5-dev $ sudo apt-get install zlib1g-dev $ wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2 $ tar jxvf samtools-1.3.1.tar.bz2 $ cd samtools-1.3.1 $ ./configure $ sudo apt-get install make $ make $ sudo make install
$ wget https://github.com/samtools/bcftools/releases/download/1.3.1/bcftools-1.3.1.tar.bz2 $ ... $ sudo apt-get install gcc $ sudo apt-get install zlib1g-dev $ make $ sudo make install
Logouts of the SSH session were required to update /usr/local/bin/samtools and bcftools after their installations.
tabix/bgzip was also useful.
$ sudo apt-get install tabix
Integrative Genomics Viewer (IGV)
written in Java and works in GUI environment
I used this software in Windows desktop environment.
Data analysis
alignment (140h)
$ ~/FastQC/fastqc -o reports -t 1 <*.fastq.gz>took about 0.5h
$ bwa index -p GRCm38_index Mus_musculus.GRCm38.74.dna.toplevel.fa.gztook about 1.5-2h
$ bwa aln -n0.02 ../../genome/GRCm38_index DRR028646_1.fastq > DRR028646_1.sai
$ bwa aln -n0.02 ../../genome/GRCm38_index DRR028646_2.fastq.gz > DRR028646_2.saitook about 50-60h each; consumed max. 5G byte; bwa aln for .fastq.bz2 didn't work in my environment for unknown reasons; .sai = 5-6G byte
took about 8h; consumed max. 7G byte; .sam = 130G byte$ nohup bwa sampe -a2000000 ../../genome/GRCm38_index DRR028646_1.sai DRR028646_2.sai DRR028646_1.fastq.gz DRR028646_2.fastq.gz > DRR028646.sam 2> stderr.log &
$ nohup bwa sampe -a2000000 ../../genome/GRCm38_index DRR028646_1.sai DRR028646_2.sai DRR028646_1.fastq.gz DRR028646_2.fastq.gz 2> stderr.log \ | samtools view -bS - > DRR028646.bam 2> stderr.2.log &took about 8.5h; consumed max. 7G byte; .bam = 44G byte
$ nohup samtools sort DRR028646.bam -o DRR028646.sort.bam &took about 2h; consumed around 1G byte; sorted .bam = 32G byte
Multithread option (e.g., "-@6") worked well in this step.
$ mv DRR028646.sort.bam DRR028646.bam $ samtools index DRR028646.bamtook about 10 min
variant calling (20h)
$ nohup samtools mpileup -uf ../../genome/Mus_musculus.GRCm38.74.dna.toplevel.fa.gz DRR028646.bam 2> stderr.log \ | bcftools call -vc -Oz > DRR028646.vcf.gz 2> stderr.2.log &took about 18h; .vcf.gz size = 320M byte
Note: the idexing of gzipped .fa.gz is not supported (only bgzip compression is supported; bgzip is included in tabix package).
Note: the usage of bcftools is very different from that of older versions. Most of the examples you can find in the web are obsolete.
$ bcftools stats DRR028646.vcf.gz > vcfstats
| accession | name | number of SNPs | number of INDELs |
|---|---|---|---|
| DRR028532 | FLS3 | 5200551 | 804857 |
| DRR028542 | CTS1 | 5210480 | 814377 |
| DRR028638 | 129/GFP ES | 5241069 | 828393 |
| DRR028644 | ntESG1 | 5018359 | 789025 |
| DRR028646 | FES1 | 5208496 | 803758 |
| DRR028650 | ntESG2 | 4942827 | 761603 |
| DRR028652 | FES2 | 5040101 | 761845 |
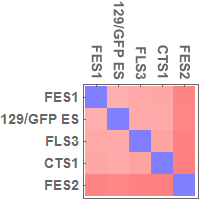
SNP analysis (1st. trial - obsolete)
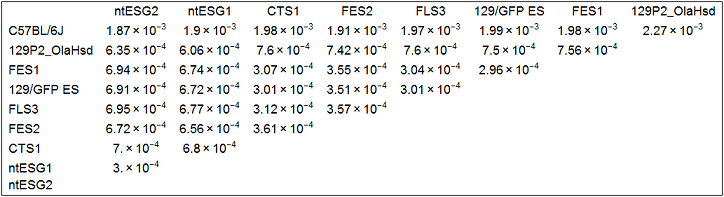
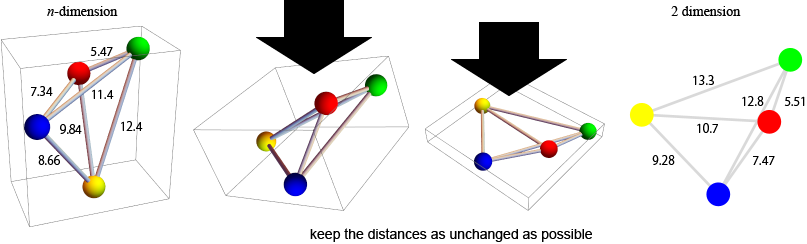
Multidimensional scaling (MDS)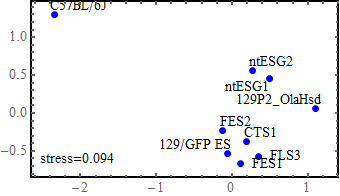
I used all the SNVs detected in the automated process, which potentially contain a lot of false positives.
Note: I found that the distance that I calculated from all the SNVs detected in FES1 replicates (DRR028646 and DRR028647) was 3.03 x 10^-4. Distances around this value threfore should be considered to indicate "background" differences among genetically identical cell lines in this analysis. For more precise comparison of similarities between the cells, it will be required to filter out a lot of false positive SNVs by using some criteria.
the idea of MDS
SNP analysis (2nd. trial)
I this 2nd. trial, I use techniques learned from my experiences on reproducing the figures of Endo-2014 to obtain more reliable results.
Extraction of SNPs that can distinguish strain 129 from B6
extract-129B6-SNPs.py by Expo70
import sys
def main():
# ouput in VCF format
col_chr = 0
col_pos = 1
col_SNPid = 2
col_ref = 3
col_alt = 4
col_qual = 5
col_filter = 6
col_info = 7
col_129P2 = 10-1
col_129S1 = 11-1
col_129S5 = 12-1
col_C57B6NJ = 17-1
col_strains = [col_C57B6NJ, col_129P2, col_129S1, col_129S5]
lineno = 0
with sys.stdin as fin: #use as a filter
for line in fin:
li = line.rstrip()
if (len(li) >= 2) and (li[0] == '#') and (li[1] == '#'):
continue
ls = li.split("\t")
lineno = lineno + 1
if lineno == 1: #check file header
labels = { col_chr:"#CHROM", col_pos:"POS", col_SNPid:"ID", col_ref:"REF", col_alt:"ALT", col_qual:"QUAL", col_filter:"FILTER", col_info:"INFO", col_129P2:"129P2", col_129S1:"129S1", col_C57B6NJ:"C57BL6NJ" }
for col in labels.keys():
if ls[col] != labels[col]:
print >> sys.stderr, "column '" + labels[col] + "' could not be found in the input VCF file"
return 2
print "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO"
else:
genotypes = [(ls[i].split(":"))[0] for i in col_strains]
if (genotypes[0]==".") or (genotypes[1]==".") or (genotypes[2]==".") or (genotypes[3]=="."):
continue
if genotypes[1]==genotypes[2]==genotypes[3]:
g0 = genotypes[0].split("/")
g1 = genotypes[1].split("/")
if (g0[0]==g0[1]) and (g1[0]==g1[1]) and (g0[0]!=g1[0]):
print "%s\t%s\t%s\t%s\t%s\t.\t.\t%s" %(ls[0],ls[1],ls[2],ls[3],ls[4],g0[0])
return 0
if __name__ == '__main__':
main()
$ cd stap/snps/dbSNP $ zcat mgp.v3.snps.rsIDdbSNPv137.vcf.gz | python ~/extract-129B6-SNPs.py > 129B6-SNPs.vcfIt extracted the information of 5030545 SNPs from a SNPs database.
129B6-SNPs.vcf.gz (44MB)
Counting alles of the extracted SNPs in .bam files
I used snpexp. For the usage and installation of this software, see my report on trisomy-8 detection.
$ cd stap/reads/FES1 $ ~/snpexp/src/snpexp -V ~/stap/snps/dbSNP/129B6-SNPs.vcf DRR028646.p.MD.bam > DRR028646.p.MD.129B6.snpexp.out 2>/dev/null
Update: I found the original version of snpexp has a bug in the routine for allele counting.
Therefore I use a fixed version snpexp2 here.
I also use the original (no extractions of properly mapped reads) .bam files in this section for simplification.
I have checked the effects of the extractions only sligtly change the resultant .bam files and there are no apparent differences in the visualized alllele distributions in the next section.
$ cd stap/reads/FES1 $ ~/snpexp/src/snpexp2 -V ~/stap/snps/dbSNP/129B6-SNPs.vcf DRR028646.bam > DRR028646.129B6.snpexp2.outDRR028646.129B6.snpexp2.out.gz (52MB)
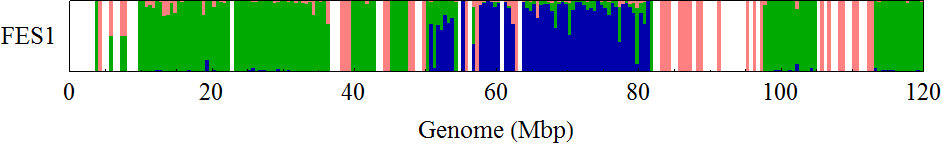
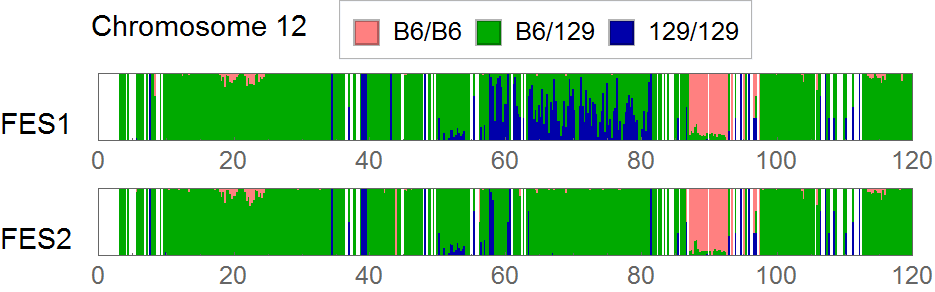
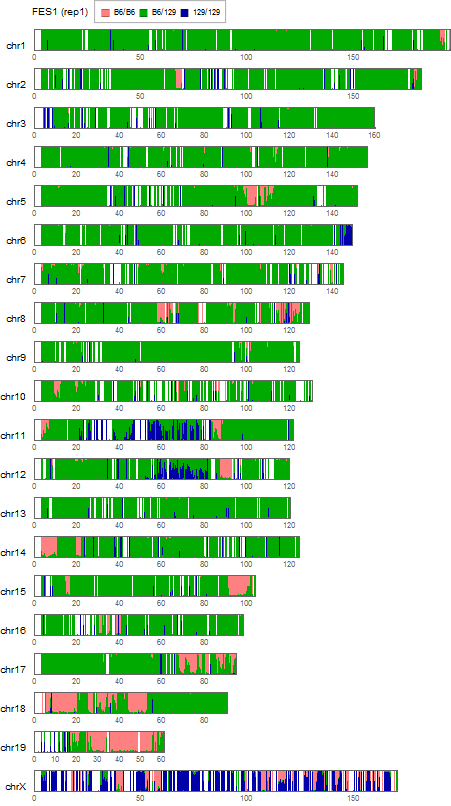
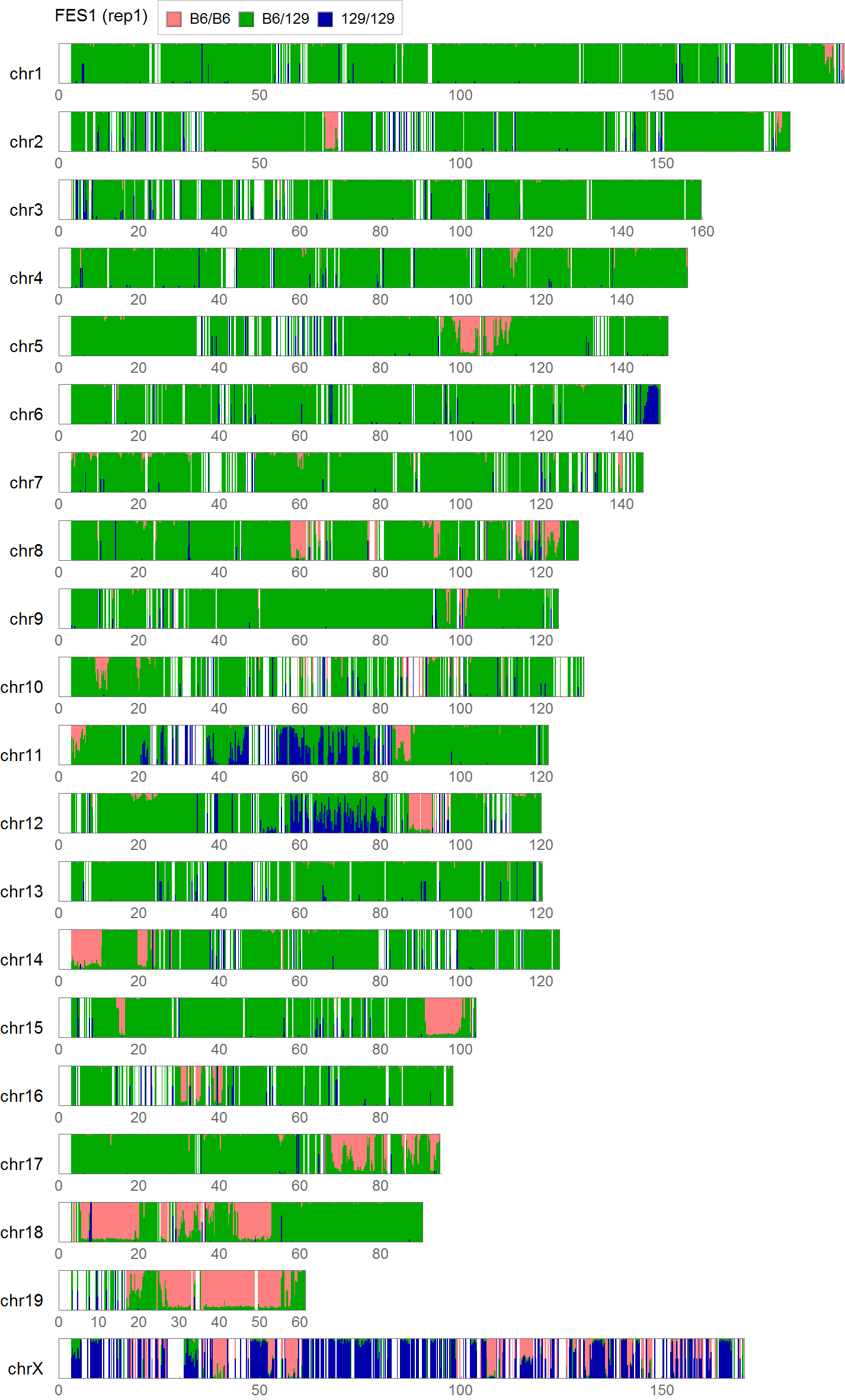
Visualization of the positional distribution of allele frequencies
visualization parameters:
- bin size = 0.25Mb
- for each SNP,
- B6/B6 homo: B6 type allele > 95%
- 129/129 homo: B6 type allele < 5%
- 129/B6 hetero: the others
{kind=link}
Allele frequency distribution for total chromosome
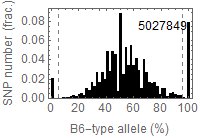
Dashed lines represent thresolds for homo/hetero discrimination.
filtering SNVs
Ref: Supplementary Information of [3]
After filtering properly mapped tags and then removing PCR duplication with SAMtools (ver. 0.1.19), we identified SNPs from the aligned sequence data using SAMtools Mpileup and BCFtools (ver. 0.1.19) with default options. We also identified reliable SNVs including heterozygous alleles from aligned sequence data that satisfied the following criteria using SAMtools Mpileup after filtering the properly mapped tags. (1) Sequence coverage > 20. (2) At least 25% of reads supported alternative alleles that differed from the B6 reference sequence.
flags in .sam/.bam file
$ samtools flags
Flags: 0x1 PAIRED .. paired-end (or multiple-segment) sequencing technology 0x2 PROPER_PAIR .. each segment properly aligned according to the aligner 0x4 UNMAP .. segment unmapped 0x8 MUNMAP .. next segment in the template unmapped 0x10 REVERSE .. SEQ is reverse complemented 0x20 MREVERSE .. SEQ of the next segment in the template is reversed 0x40 READ1 .. the first segment in the template 0x80 READ2 .. the last segment in the template 0x100 SECONDARY .. secondary alignment 0x200 QCFAIL .. not passing quality controls 0x400 DUP .. PCR or optical duplicate 0x800 SUPPLEMENTARY .. supplementary alignment
| 0x | 8 | 4 | 2 | 1 | 8 | 4 | 2 | 1 | 8 | 4 | 2 | 1 |
| S | d | f | s | 2 | 1 | R | r | U | u | P | p | |
| prorperly paired | 0 | 1 | 1 | |||||||||
| with itself and mate mapped | 0 | 0 | 1 | |||||||||
| singletons | 1 | 0 | 1 |
I extracted properly paired reads.
$ samtools view -b -f 0x02 DRR028646.bam > DRR028646.p.bamtook 1h; from DRR028646.bam (32GB), got DRR028646.p.bam (28GB)
For the removal of potential PCR duplicates, I used picard MarkDuplicates instead of samtools rmdup.
$ cd $ wget -c https://github.com/broadinstitute/picard/releases/download/2.6.0/picard.jar $ cd stap/reads/FES1 $ java -jar ~/picard.jar MarkDuplicates I=DRR028646.p.bam O=DRR028646.p.MD.bam M=DRR028646.picard_metrics.txt VALIDATION_STRINGENCY=SILENTtook 2h; got DRR028646.p.MD (29GB)
By using samtools mpileup, I got DRR028646.p.MD.vcf.gz.
I used an automation script get_vcf2.sh in the real processings.
$ cd $ nohup ./get_vcf2.sh DRR028646 & $ nohup ./get_vcf2.sh DRR028647 &
"Reliable SNVs"
I obtained the list of reliable SNPs in FES1 data by using a script
extract-reliable-SNPs.py by Expo70
import sys
def main():
# usage $python extract-reliable-SNPs.py [command]
# ouput in VCF format
min_allele_ratio = 0.25
min_depth = 20+1
is_skip_counts = False
if len(sys.argv) == 2:
if sys.argv[1] == "ignore-allele-ratio":
min_allele_ratio = 0.0
elif sys.argv[1] == "skip-counts":
is_skip_counts = True
else:
print >> sys.stderr, "uknown command, neither of\n\tignore-allele-ratio\n\tskip-counts"
return 3
col_chr = 0
col_pos = 1
col_SNPid = 2
col_ref = 3
col_alt = 4
col_qual = 5
col_filter = 6
col_info = 7
lineno = 0
with sys.stdin as fin: #use as a filter
for line in fin:
li = line.rstrip()
if (len(li) >= 2) and (li[0] == '#') and (li[1] == '#'):
continue
ls = li.split("\t")
lineno = lineno + 1
if lineno == 1: #check file header
labels = { col_chr:"#CHROM", col_pos:"POS", col_SNPid:"ID", col_ref:"REF", col_alt:"ALT", col_qual:"QUAL", col_filter:"FILTER", col_info:"INFO" }
for col in labels.keys():
if ls[col] != labels[col]:
print >> sys.stderr, "column '" + labels[col] + "' could not be found in the input VCF file"
return 2
print "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO"
else:
info = ls[col_info].split(";")
depth = 0
ref_count = 0
alt_count = 0
alt_allele_ratio = 0.0
for i in info:
if i == "INDEL":
break
key_value = i.split("=")
if key_value[0] == "DP":
depth = int(key_value[1])
elif key_value[0] == "DP4":
counts = key_value[1].split(",")
ref_count = int(counts[0])+int(counts[1])
alt_count = int(counts[2])+int(counts[3])
if depth != 0:
alt_allele_ratio = float(alt_count)/depth
if (depth >= min_depth) and (alt_allele_ratio >= min_allele_ratio):
#print li
if is_skip_counts:
print "%s\t%s\t%s\t%s\t%s\t.\t.\t." %(ls[0],ls[1],ls[2],ls[3],ls[4])
else:
print "%s\t%s\t%s\t%s\t%s\t.\t.\t%d,%d" %(ls[0],ls[1],ls[2],ls[3],ls[4], ref_count,alt_count)
return 0
if __name__ == '__main__':
main()
$ zcat DRR028646.p.MD.vcf.gz | python ~/extract-reliable-SNPs.py skip-counts > DRR028646.p.MD.erSNPs.vcf $ zcat DRR028647.p.MD.vcf.gz | python ~/extract-reliable-SNPs.py skip-counts > DRR028647.p.MD.erSNPs.vcf $ cat DRR028646.p.MD.erSNPs.vcf DRR028647.p.MD.erSNPs.vcf | LC_COLLATE=C sort -u > temp $ grep '^#' temp > FES1.erSNPs.vcf && grep -v '^#' temp | LC_ALL=C sort -t ' ' -k1,1 -k2,2n >> FES1.erSNPs.vcfNote: Input ' ' as a single quote, CTRL-V and then TAB, a single quote in the command shell.
$ grep -v "^#" FES1.erSNPs.vcf | wc -l
780088
extracting reliable SNVs in FES1 that are not observed in FES2
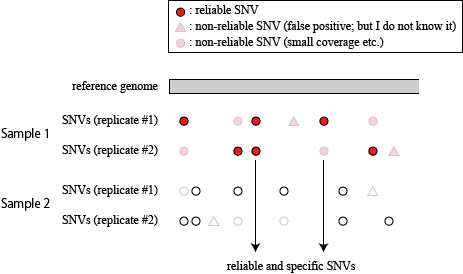
From now on, I will use a bug-fixed version of snpexp (snpexp2).
This fixed version is essential to obtain precise results in this section.
extract-negatives.py by Expo70
import sys, getopt
try:
opts, args = getopt.getopt(sys.argv[1:], "m:")
except getopt.GetoptError as err:
print str(err)
sys.exit(1)
if len(args)!=1:
sys.exit(2)
filename = args[0]
base_pos = { "A":0, "C":1, "G":2, "T":3 }
min_total_count = 1
for o,a in opts:
if o=="-m":
if a.isdigit():
min_total_count = int(a)
else:
print >> sys.stderr, "argument for -m option should be non-negative integer"
sys.exit(3)
else:
assert False, "undefined option"
with open(filename, "r") as fin:
print "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO"
for line in fin:
li = line.rstrip()
ls = li.split("\t")
alt = ls[5-1].split(",")
acgt = ls[6-1].split(",")
if len(acgt) == 4:
alt_count = 0
for a in alt:
alt_count = alt_count + int(acgt[base_pos[a]])
if alt_count == 0:
total_count = 0
for i in [0,1,2,3]:
total_count = total_count + int(acgt[i])
if total_count >= min_total_count:
#print li
print "%s\t%s\t%s\t%s\t%s\t.\t.\t." %(ls[0],ls[1],ls[2],ls[3],ls[4])
fix-und.sh by Expo70
#!/bin/bash set -e set -u set -o pipefail grep -v "undetermined" $1 > $1.temp mv $1.temp $1 exit 0
$ ~/snpexp/src/snpexp2 -V ../FES1/FES1.erSNPs.vcf DRR028652.bam > DRR028652.FES1.erSNPs.snpexp2.out $ ~/snpexp/src/snpexp2 -V ../FES1/FES1.erSNPs.vcf DRR028653.bam > DRR028653.FES1.erSNPs.snpexp2.out $ ./fix-und.sh DRR028652.FES1.erSNPs.snpexp2.out $ ./fix-und.sh DRR028653.FES1.erSNPs.snpexp2.out $ python ~/extract-negatives.py DRR028652.FES1.erSNPs.snpexp2.out > DRR028652.-FES1.erSNPs.vcf $ python ~/extract-negatives.py DRR028653.FES1.erSNPs.snpexp2.out > DRR028653.-FES1.erSNPs.vcf $ comm -1 -2 <(sort DRR028652.-FES1.erSNPs.vcf) <(sort DRR028653.-FES1.erSNPs.vcf) > temp $ grep "^#" temp > FES2-FES1.erSNPs.vcf; grep -v "^#" temp | LC_ALL=C sort -t ' ' -k1,1 -k2,2n >> FES2-FES1.erSNPs.vcf
fix-und.sh is required to remove trailing garbases in the outputs that are often added when snpexp causes segmentation fault.
This process is not required after fixing this problem.
$ grep -v "^#" FES2-FES1.erSNPs.vcf | wc -l $ cut -f1 FES2-FES1.erSNPs.vcf | uniq -c
14147
1 #CHROM
11 1
11 10
8889 11
4541 12
14 13
1 14
6 15
6 16
10 17
7 18
3 19
12 2
9 3
14 4
18 5
567 6
13 7
7 8
6 9
1 X
1 Y
reverse case: reliable SNVs in FES2 that are not observed in FES1
Starting from the extraction of reliable SNVs in FES2, I obtained unique SNVs for FES2 side.
$ grep -v "^#" FES2.erSNPs.vcf | wc -l
455928
$ grep -v "^#" FES1-FES2.erSNPs.vcf | wc -l $ cut -f1 FES1-FES2.erSNPs.vcf | uniq -c
135
1 #CHROM
15 1
7 10
9 11
8 12
11 13
8 14
3 15
1 16
5 17
3 18
1 19
9 2
4 3
3 4
14 5
4 6
9 7
8 8
9 9
4 Y
Presence of FES1/FES2-specific SNVs in the STAP-related cells
Note: I fixed an additional minor bug in snpexp. I used this fixed version in this section.
$ ~/snpexp/src/snpexp2 -V ../FES2/FES2-FES1.erSNPs.vcf DRR028632.bam > DRR028632.FES2-1.erSNPs.snpexp2.out & $ ~/snpexp/src/snpexp2 -V ../FES1/FES1-FES2.erSNPs.vcf DRR028632.bam > DRR028632.FES1-2.erSNPs.snpexp2.out &
Extracting SNVs that exists in one of FES cell lines but not in the other can remove the effect of genetic background common to the parental mice used to establish these ES cells. But the genome-wide comparison between FES1 and FES2 SNPs distributions has reveled the presence of FES1-specific 129/129-homo clusters in some of the chromosomes [ref1,3]. This suggests that heterogeneity in the chromosomal level exists in the parental B6 mice and is differentially inherited in FES cell lines. To remove SNVs that are considered to be originated from this chromosomal event, I extracted SNVs in regions other than chromosome 6, 11, and 12 where the FES1-specific 129/129-home clusters are observed.
$ grep -v -E '^6|^11|^12' DRR028632.FES2-1.erSNPs.snpexp2.out > DRR028632.FES1-SNV.snpexp2.out $ grep -v -E '^6|^11|^12' DRR028632.FES1-2.erSNPs.snpexp2.out > DRR028632.FES2-SNV.snpexp2.out
$ grep -v "^#" DRR028632.FES1-SNV.snpexp2.out | wc -l $ grep -v "^#" DRR028632.FES2-SNV.snpexp2.out | wc -l
150 114
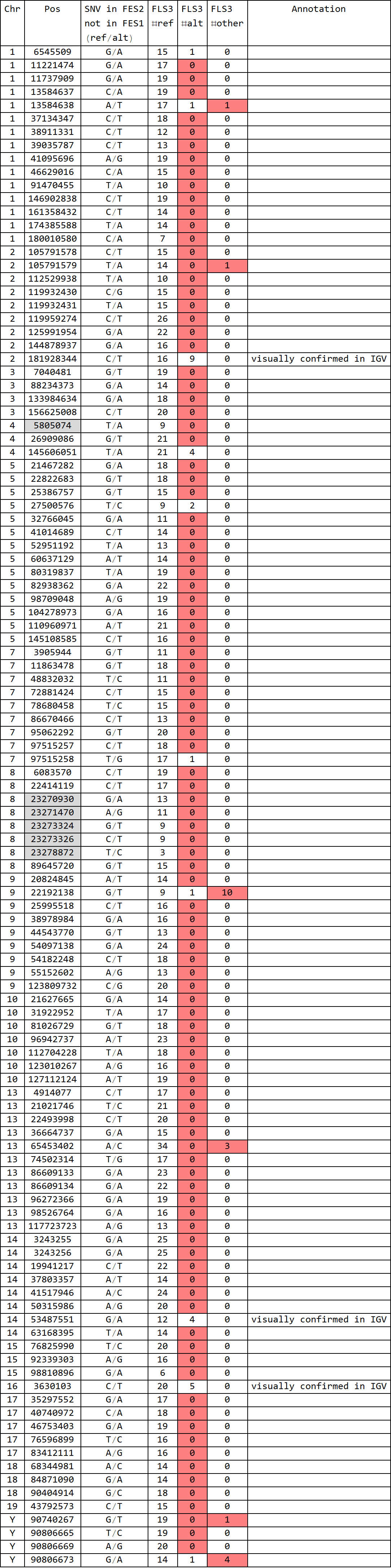
for FLS3 (rep1):
- Formatted output of DRR028632.FES1-SNV.snpexp2.out
- total 150 SNVs; 5 known SNPs in 129 v.s. B6; 8 mismatches
- final match = 137/145 (94%)
- Formatted output of DRR028632.FES2-SNV.snpexp2.out
- total 114 SNVs; 6 known SNPs in 129 v.s. B6; 3 matches
- final match = 3/108 (3%)
{kind=link}
{kind=link}
 full resolution
full resolution
{kind=link}
Data analysis environment
- Intel(R) Xeon(R) CPU E5-2650 v2 @ 2.60GHz, x6
- 8G memory
- 200G SSD
- Ubuntu 16.04 LTS
Can mutation accumulation explain genome difference between the STAP related cells and the ES cells?
...Reference