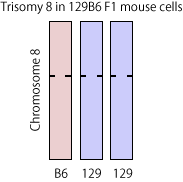
Introduction
Trisomy-8 detected in published RNA-seq data from STAP cells
The pellets of STAP related cells were brought to the NGS facility in RIKEN CDB by Ms. Obokata [1,2] and the RNA-seq/ChIP-seq analysis results from her cells were registered to the public database. Dr. Endo, through bioinformatic technique, recovered genomic SNPs from the published RNA-seq data and obtained the distribution of allele frequency in SNPs in these cells. He found the shift of the frequency distribution peak only in chromosome 8 [3] and up-regulated mRNA expressions from the same chromosome [3,4] in the samples from "STAP cells". These results indicated that "STAP cells" used in RNA-seq in STAP papers have trisomy-8, which is one of the most frequent aneuploidity in ES cells.
Although trisomy-8 was not detected in 129B6F1 cells analyzed by RIKEN scientific investigation team [1], his finding was one of the first scientific indications that STAP cells may be contaminated by ES cells. Here I just tried to reproduce a part of his results to convince myself.
Note: I am a beginner for NGS data analysis (about 2 month experience). The result presented in this page is only preliminary and in non-prefessional quality.
1st. trial
SRA110029 (ftp)
RunsIn this analysis, I used RNA-seq data genrated using SMARTer reagent kit as described in Endo-2014.
$ wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA110/SRA110029/SRX472647/SRR1171578_1.fastq.bz2 $ wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA110/SRA110029/SRX472647/SRR1171578_2.fastq.bz2 $ bzip2 -d SRR1171578_1.fastq.bz2 $ gzip SRR1171578_1.fastq $ bzip2 -d SRR1171578_2.fastq.bz2 $ gzip SRR1171578_2.fastq
- SRR1171578_1.fastq.gz (2.9G byte)
- SRR1171578_2.fastq.gz (2.7G byte)
Reference genome
You can obtain reference genome data prepared for Bowtie2 from iGenome data bank provided by Illumina.iGenome iGenome
$ cd ../../genome $ wget -c ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Mus_musculus/NCBI/GRCm38/Mus_musculus_NCBI_GRCm38.tar.gz $ tar xzvf Mus_musculus_NCBI_GRCm38.tar.gzYou will not need all the files in the archive. You may have to erase some of the files to spare the storage space.
Mapping
Since RNA sequences are modified by RNA splicing and thus different from the original genomic sequences, tools designed for the mapping of genomic reads such asBWA cannot be used for the mapping of RNA reads.
For this specialized purpose, you have to use splicing-aware mappers such as TopHat which predicts exon-exon junctions in the reads and revserse the process of RNA splicing in silico.
TopHat
maps short sequences from spliced transcripts to whole genomes.originally developed by Cole Trapnell at the University of Maryland
written in Python and C++
internally uses
Bowtie aligner and samtools v.0.1.18
$ sudo apt-get install tophat $ tophat -vI used v.2.1.0
#!/bin/bash
set -e
set -u
set -o pipefail
id="SRR1171578"
ref="../../genome/Mus_musculus/NCBI/GRCm38/Sequence/Bowtie2Index/genome"
gtf="../../genome/Mus_musculus/NCBI/GRCm38/Annotation/Genes/genes.gtf"
cpu_cores=1
tophat2 -o ${id}.tophat -p ${cpu_cores} -no-coverage-search -G ${gtf} ${ref} ${id}_1.fastq.gz ${id}_2.fastq.gz
It took about 85h and consumed maximally about 3G-byte memory when using 1 CPU core.
I reproducibily got errors near the final stage of the mapping when using 3 CPU cores in my environment probably due to the insufficient memory.
In those cases, resuming by using -R option didn't work because of the loss of some data.
Finally, I got 6 output files:
- align_summary.txt
- accepted_hits.bam
- junctions.bed
- insertions.bed
- deletions.bed
- unmapped.bam
| Accesion | Name | accepted_hits.bam | Index of .bam | junctions.bed | insertions.bed | deletions.bed | align_summary.txt |
|---|---|---|---|---|---|---|---|
| SRR1171578 | STAP-rep1 | SRR1171578.bam (3.3GB) , stats. | SRR1171578.bam.bai (2MB) | SRR1171578-j.bed (6MB) | SRR1171578-i.bed (0.6MB) | SRR1171578-d.bed (2MB) | align_summary.txt |
| SRR1171579 | STAP-rep2 | SRR1171579.bam (3.4GB) , stats. | SRR1171579.bam.bai (2MB) | SRR1171579-j.bed (6MB) | SRR1171579-i.bed (0.7MB) | SRR1171579-d.bed (2MB) | align_summary.txt |
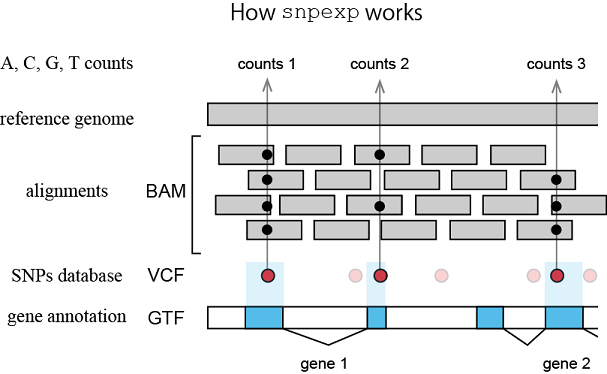
Variant calling and counting
from Endo-2014 [3]:Sequence fragments aligned on the mouse genome were analyzed using the SNP detection and enumeration program mentioned above [snpexp], and only SNPs with cover ratios ≥20 were retained. If ≥95% of the SNP sequence was the same on the two strands, the allele was designated as homozygous. Genomewide heterozygosity between B6 and 129 was analyzed using a subset of the SNPs described above [SNPs inside exons] with different alleles between B6 and 129.[]: added by Expo70.
snpexp by Takaho Endo
Allele frequency counter with BAM file and VCF file
$ sudo apt-get install git $ git clone https://github.com/takaho/snpexp.git
$ sudo apt-get autoconf $ cd snpexp/src $ autoheader
Note that without autoconf you will get a compilation error: config.status: error: cannot find input file: `config.h.in'. Before start building snpexp, you have to build and install samtools (and maybe htslib). Unfortunately, snpexp seems to be based on older (pre 0.1.19) versions of samtools. If you are using the latest version of samtools, the source codes require some modifications:
- from samtools archive, copy bam.h and htslib/bgzf.h into snpexp/src directory.
-
add an include directive below to src/snpexp.cxx
#include "config.h" -
add this to src/config.h
#define HAVE_BAM_H 1 -
modify src/Makefile.in
LIB = -lz -lbam -lpthread
to
LIB = -lz -lhts -lpthread
$ ./configure $ make
$ wget -c ftp://ftp.ncbi.nih.gov/snp/organisms/mouse_10090/VCF/genotype/SC_MOUSE_GENOMES.genotype.vcf.gz $ gzip -d -k SC_MOUSE_GENOMES.genotype.vcf.gz
$ ~/snpexp/src/snpexp -o snpexp.out -H \ -G ~/stap/genome/Mus_musculus/NCBI/GRCm38/Annotation/Genes/genes.gtf \ -V ~/stap/snps/dbSNP/SC_MOUSE_GENOMES.genotype.vcf \ -m 20 -s C57BL/6NJ,129P2/OlaHsd,129S1/SvImJ,129S5SvEv SRR1171578.bamNote:
snpexp does not support .vcf.gz files. Use .vcf files.
af.py (extracts SNPs that differ between 129 and the reference B6)
written by Expo70
import sys
filename = sys.argv[1]
with open(filename, "r") as fin:
for line in fin:
li = line.rstrip()
ls = li.split("\t")
ref = ls[4-1]
alt = ls[5-1]
strains = ls[6-1]
b6A = strains[0:2]
s129A = strains[2:4]
s129B = strains[4:6]
s129C = strains[6:8]
acgt = ls[7-1].split(",")
if len(acgt) == 4:
# if (alt in s129A) and (alt in s129B) and (alt in s129C) and not (alt in b6A):
if (alt in s129A) and (alt in s129B) and (alt in s129C):
print li
$ python af.py snpexp.out > snpexp.129.out
Allele frequencies
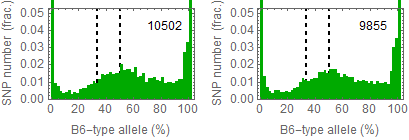
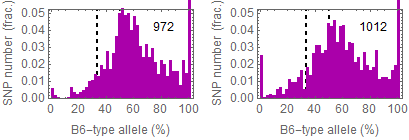
Ref: Endo-2014, Fig.3A
Allele frequencies of SNPs in total chromosome (upper) and chromosome 8 (lower)
Allele frequencies in total chromosome (STAP-rep1, STAP-rep2) obtained in my analysis; numbers in the frames indicate the counts of SNPs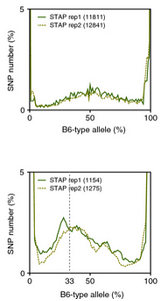
In this 1st. trial, I calculated B6-type allele frequencies as [the numbers of the reads that have reference base]/[the total numbers of the reads] for each extracted SNP site in snpexp.129.out.
I created histograms by setting the numbers of bins 10.
The obtained distributions and the numbers of SNPs counted were slightly different from those in the original paper.
This is probably due to my misunderstandings of the ways of extracting SNPs described by the author.
Nevertheless, I could reproduce allele frequency distributions whose peaks were around 33% B6-type allele in chromosome 8 as shown in the original paper.
mRNA expressions
| Accesion | Name | accepted_hits.bam | Index of .bam | junctions.bed | insertions.bed | deletions.bed | align_summary.txt |
|---|---|---|---|---|---|---|---|
| SRR1171574 | ESC-rep1 | SRR1171574.bam (3.3GB) , stats. | SRR1171574.bam.bai (2MB) | SRR1171574-j.bed (6MB) | SRR1171574-i.bed (0.6MB) | SRR1171574-d.bed (2MB) | align_summary.txt |
| SRR1171575 | ESC-rep2 | SRR1171575.bam (3.8GB) , stats. | SRR1171575.bam.bai (2MB) | SRR1171575-j.bed (6MB) | SRR1171575-i.bed (0.6MB) | SRR1171575-d.bed (2MB) | align_summary.txt |
$ wget -c http://cole-trapnell-lab.github.io/cufflinks/assets/downloads/cufflinks-2.2.1.Linux_x86_64.tar.gz $ tar xvf cufflinks-2.2.1.Linux_x86_64.tar.gz
$ ~/cufflinks-2.2.1.Linux_x86_64/cuffdiff -o output.cuffdiff \ ../genome/Mus_musculus/NCBI/GRCm38/Annotation/Genes/genes.gtf \ ESC-rep1/SRR1171574.tophat/SRR1171574.bam,ESC-rep2/SRR1171575.tophat/SRR1171575.bam \ STAP-rep1/SRR1171578.tophat/SRR1171578.bam,STAP-rep2/SRR1171579.tophat/SRR1171579.bam
It took about 2.5h to obtain output.cuffidiff/gene_exp.diff.
gene_exp.diff (2MB) was plotted by using trisomy.R provided by Dr. Endo in RStudio in a Windows environment.
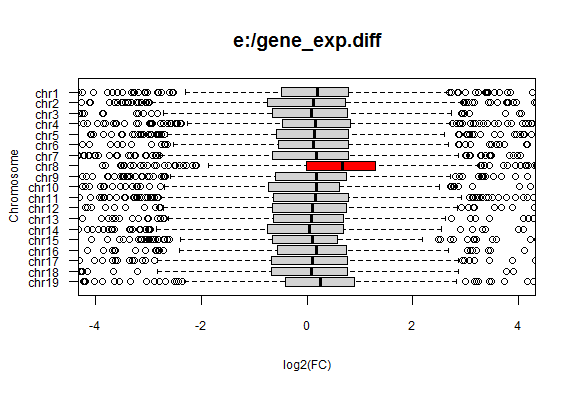
The expressions of chromosome 8 genes in "STAP" samples were 1.49x higher than those in "ESC" samples. The difference in the expressions was statistically significant only in chromosome 8 (P = 2.147e-16).
2nd. trial
Allele frequencies
afV2.py (extracts SNPs that differ between 129s and B6NJ)
written by Expo70
import sys
filename = sys.argv[1]
with open(filename, "r") as fin:
for line in fin:
li = line.rstrip()
ls = li.split("\t")
strains = ls[6-1]
sB6 = set([ strains[0], strains[1] ])
sB6.discard('*')
s129A = [ strains[2], strains[3] ]
s129B = [ strains[4], strains[5] ]
s129C = [ strains[6], strains[7] ]
s129 = set(s129A + s129B + s129C)
s129.discard('*')
acgt = ls[7-1].split(",")
if len(acgt) == 4:
if (len(sB6)==1) and (len(s129)==1) and (sB6!=s129):
print li
$ ~/snpexp/src/snpexp -o snpexp.out \ -G ~/stap/genome/Mus_musculus/NCBI/GRCm38/Annotation/Genes/genes.gtf \ -V ~/stap/snps/dbSNP/SC_MOUSE_GENOMES.genotype.vcf \ -m 20 -s C57BL/6NJ,129P2/OlaHsd,129S1/SvImJ,129S5SvEv SRR1171578.bam $ python afV2.py snpexp.out > snpexp.129B6.out
Note that I didn't use -H option for snpexp in this 2nd. trial.
3rd. trial
In this trial, I used an updated (?; REL-1303) version of dbSNP file. This file was quite large when uncompressed (around 40GB).
$ wget -c ftp://ftp-mouse.sanger.ac.uk/REL-1303-SNPs_Indels-GRCm38/mgp.v3.snps.rsIDdbSNPv137.vcf.gz $ gzip -d -k mgp.v3.snps.rsIDdbSNPv137.vcf.gz
$ ~/snpexp/src/snpexp -o snpexp.out \ -G ~/stap/genome/Mus_musculus/NCBI/GRCm38/Annotation/Genes/genes.gtf \ -V ~/stap/snps/dbSNP/mgp.v3.snps.rsIDdbSNPv137.vcf \ -m 20 -s C57BL6NJ,129P2,129S1,129S5 SRR1171578.bam $ python afV2.py snpexp.out > snpexp.129B6.out
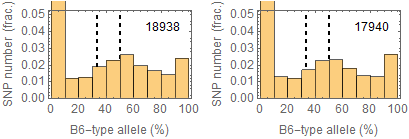
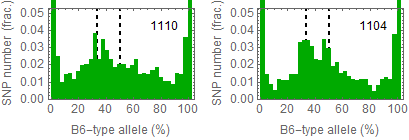
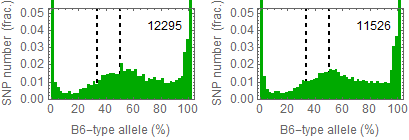
Allele frequencies
Ref: Endo-2014, Fig.3A
Allele frequencies of SNPs in total chromosome (upper) and chromosome 8 (lower)
for "CD45+ cell" (right) and "STAP cell" (right)
Allele frequencies in total chromosome (STAP-rep1, STAP-rep2) obtained in my analysis; numbers in the frames indicate the counts of SNPs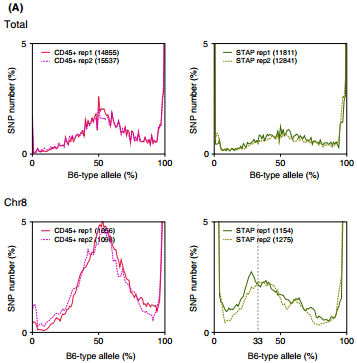
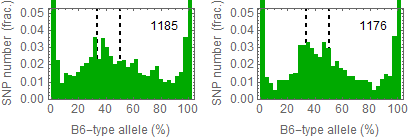
4th. trial
a bug in snpexp
I found an apparent bug in snpexp. The software does not proerly handle CIGAR strings for insertions (BAM_CIN) and deletions (BAM_CDEL); It handles them as opposite meanings. I fixed this problem as below. I confirmed that the original allele counts somtimes did not match the counts observed in IGV, but did match after the fixation. This means that the allele frequencies obtained by the original version are slightly different from the expected values. I also added the support for soft clips (BAM_CSOFT_CLIP), which seem to be often used in alignments by bwa.
from the original source code (snpexp/src/snpexp.cxx)
The MIT Lincense
Copyright (c) 2014 Takaho A. Endo
void base_frequency::add(int pos, const bam1_t* read) {
clear_reference();
// check cigar
int len = read->core.n_cigar;
#ifdef DEBUG
if (len < 4) return;
#endif
int seqpos = 0;
const uint32_t* cigar = bam1_cigar(read);
const uint8_t* sequence = bam1_seq(read);
int offset = 4;
for (int i = 0; i < len; i++) {
int op = bam_cigar_op(cigar[i]);
int slen = bam_cigar_oplen(cigar[i]);
#ifdef DEBUG
cout << pos << "\t";
cout << op << "/" << slen << " ";
#endif
if (slen + pos >= _length) {
slen = _length - pos;
}
//char c = '\0';
if (op == BAM_CMATCH || op == BAM_CEQUAL || op == BAM_CDIFF) {
for (int j = 0; j < slen; j++) {
uint8_t base = (sequence[(j+seqpos) >> 1] >> offset) & 15;
offset = 4 - offset;
switch (base) {
case 15: break; // N
#ifdef DEBUG
case 1: _a[pos + j] ++; cout << "A"; break;
case 2: _c[pos + j] ++; cout << "C"; break;
case 4: _g[pos + j] ++; cout << "G"; break;
case 8: _t[pos + j] ++; cout << "T"; break;
#else
case 1: _a[pos + j] ++; ; break;
case 2: _c[pos + j] ++; ; break;
case 4: _g[pos + j] ++; ; break;
case 8: _t[pos + j] ++; ; break;
#endif
}
}
#ifdef DEBUG
cout << " ";
#endif
pos += slen;
seqpos += slen;
} else if (op == BAM_CINS || op == BAM_CREF_SKIP) {
#ifdef DEBUG
cout << "=" << len << ">";
#endif
pos += slen;
} else if (op == BAM_CDEL) {
break;
} else {
cerr << "unknown cigar character " << op << endl;
}
}
#ifdef DEBUG
cout << endl;
#endif
}
fixed source code
void base_frequency::add(int pos, const bam1_t* read) {
clear_reference();
// check cigar
int len = read->core.n_cigar;
#ifdef DEBUG
if (len < 4) return;
#endif
int seqpos = 0;
const uint32_t* cigar = bam1_cigar(read);
const uint8_t* sequence = bam1_seq(read);
int offset = 4;
for (int i = 0; i < len; i++) {
int op = bam_cigar_op(cigar[i]);
int slen = bam_cigar_oplen(cigar[i]);
#ifdef DEBUG
cout << pos << "\t";
cout << op << "/" << slen << " ";
#endif
if (slen + pos >= _length) {
slen = _length - pos;
}
//char c = '\0';
if (op == BAM_CMATCH || op == BAM_CEQUAL || op == BAM_CDIFF) {
for (int j = 0; j < slen; j++) {
uint8_t base = (sequence[(j+seqpos) >> 1] >> offset) & 15;
offset = 4 - offset;
switch (base) {
case 15: break; // N
#ifdef DEBUG
case 1: _a[pos + j] ++; cout << "A"; break;
case 2: _c[pos + j] ++; cout << "C"; break;
case 4: _g[pos + j] ++; cout << "G"; break;
case 8: _t[pos + j] ++; cout << "T"; break;
#else
case 1: _a[pos + j] ++; ; break;
case 2: _c[pos + j] ++; ; break;
case 4: _g[pos + j] ++; ; break;
case 8: _t[pos + j] ++; ; break;
#endif
}
}
#ifdef DEBUG
cout << " ";
#endif
pos += slen;
seqpos += slen;
} else if (op == BAM_CDEL || op == BAM_CREF_SKIP) {
#ifdef DEBUG
cout << "=" << len << ">";
#endif
pos += slen;
} else if (op == BAM_CINS || op == BAM_CSOFT_CLIP) {
for (int j = 0; j < slen; j++) {
offset = 4 - offset;
}
seqpos += slen;
} else {
cerr << "unknown cigar character " << op << endl;
}
}
#ifdef DEBUG
cout << endl;
#endif
}
snpexp2 in this site.
minor bugs
chromosome M problem
snpexp treats the names of chromosome that start with "M" as chrM (mitochondorial genome). But GRCm38 reference genome contains MG3835_PATCH, which is not chrM.
from the original source code (snpexp/src/tktools.cxx)
int tktools::bio::convert_chromosome_to_code(const char* name) {
int code;
if (strncmp(name, "chr", 3) == 0) {
name += 3;
}
if ( name[ 0 ] == 'X' ) {//strcmp( name, "X" ) == 0 ) {
code = 64;
} else if ( name[ 0 ] == 'Y' ) {// strcmp( name, "Y" ) == 0 ) {
code = 65;
} else if ( name[ 0 ] == 'W' ) {
code = 66;
} else if ( name[ 0 ] == 'Z' ) {
code = 67;
} else if ( name[ 0 ] == 'M' || strcmp( name, "mitochondria" ) == 0 ) {
code = 128;
fixed source code
int tktools::bio::convert_chromosome_to_code(const char* name) {
int code;
if (strncmp(name, "chr", 3) == 0) {
name += 3;
}
if ( name[ 0 ] == 'X' ) {//strcmp( name, "X" ) == 0 ) {
code = 64;
} else if ( name[ 0 ] == 'Y' ) {// strcmp( name, "Y" ) == 0 ) {
code = 65;
} else if ( name[ 0 ] == 'W' ) {
code = 66;
} else if ( name[ 0 ] == 'Z' ) {
code = 67;
} else if ( std::string(name)=="M" || std::string(name)=="MT" || strcmp( name, "mitochondria" ) == 0 ) {
code = 128;
segmentation fault problem
In some cases, depending on input .bam files, snpexp causes segmentation fault when it accesses an array target_name[] when its index is equal to -1. This problem could be fixed by adding a line to the souce code as below:
//add this line to snpexp/src/snpexp.cxx, around line 800
if(current_chromosome==-1)break;
// read
bool flag_skipping = false;
for (int i = 0; i < num_bams; i++) {
if (chromosomes[i] != current_chromosome) {
bfreqs[i] = NULL;
continue;
}
string chromosome_name = headers[i]->target_name[chromosomes[i]];
get_snps() range problem (added at 2017/2/13)
The output range of get_snps() is defined as [index_start, index_end), but in the final "if" clause, it is calculated as index_end]. This makes a SNP at the end of each chromosome missing. I fixed this problem:
const snp_allele* post = snps[center + 1];
//cerr << center << ":" << post->chromosome_code() << endl;
if (post->chromosome_code() != ccode) {
index_end = center + 1; // "+1" was added
int ccode = convert_chromosome_to_code(chrname.c_str());
// added a support for undetemined chromosomes (ccode == 0)
if(ccode == 0){ return make_pair(0,0); }
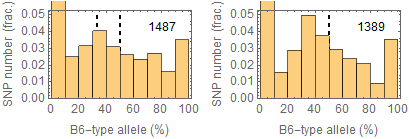
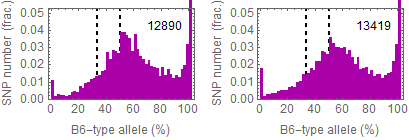
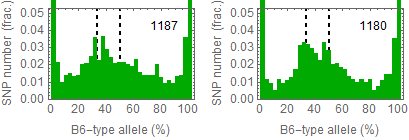
Updated results after the fixation
Allele frequencies in total chromosome (STAP-rep1, STAP-rep2) obtained in my analysis; numbers in the frames indicate the counts of SNPs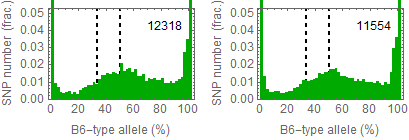
Reference
- Katsura report (J)(E)
- RIKEN press conference at 6/12/2014 (J)
- Endo 2014, Genes to Cells, "Quality control method for RNA-seq using single nucleotide polymorphism allele frequency"
- 次世代シーケンサーDRY解析, "R+trisomy.R", pp.348-351